A company of consultants in chemistry and chemical engineering, actively involved in computational, experimental, and environmental organic chemistry.
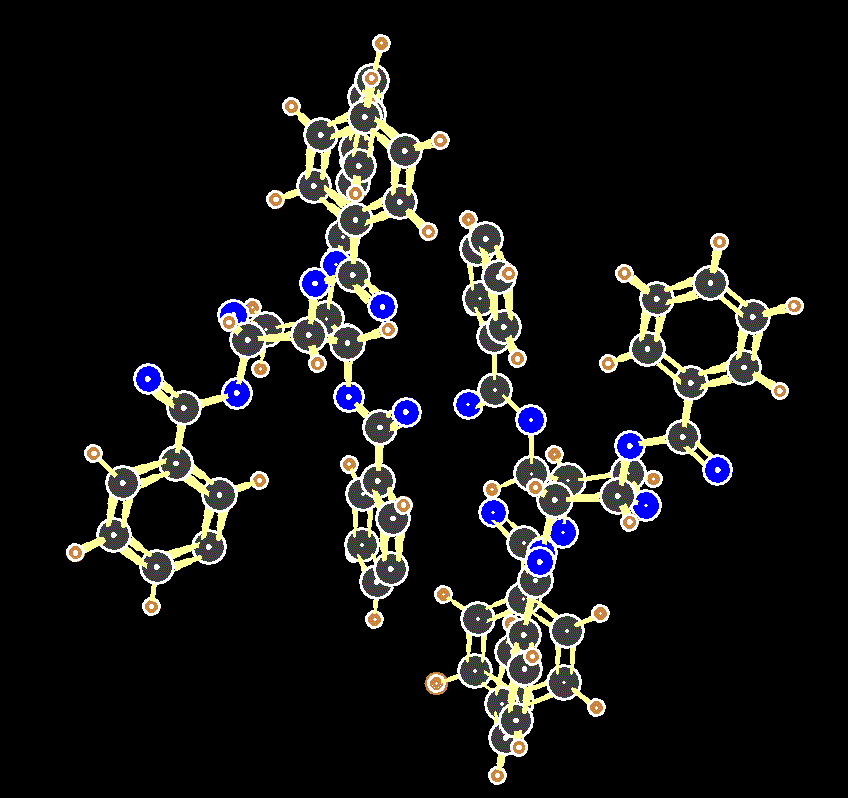
Two molecules of the tetrabenzoate of xylopyranose in the unit cell of its crystal. Note that the counter-intuitive axial conformation is favoured in the solid phase.
Find out about StruMM3D's molecular models, how you can generate them, and how you can use them in your daily tasks, presentations, reports and publications.
THE StruMM3D MOLECULAR MODELERS
You only purchase a license to use StruMM3D, or its utilities, once! All subsequent upgrades are FREE! Currently registered users of the StruMM3D MOLECULAR MODELER can get their free upgrade by e-mail. Simply send us your name, StruMM3D version number, and your e-mail address, and we'll send the upgrade back by e-mail. Upgrades are also available online.
The file conversion utility, which will convert most structure data files, including CIF, PDB, MDL, Schakal, XYZ, MOL, and many other structural coordinate data file types, into the very compact STR files, has been updated and fully integrated with StruMM3D.
We want to get StruMM3D to as many users as is possible, but we do want it to be as bug free and user-friendly as is possible. So, if you have used StruMM3D,then we need your frank impressions of the program. We beg you to spend a few minutes to send us e-mail with your criticisms and suggestions.
Users of the StruMM3D molecular modelers should visit the StruMM3D Tips and Shortcuts site frequently. Questions about the features and uses of the molecular modelers are answered and discussed here. Valuable tips for maximizing your efficiency when using the StruMM3D MOLECULAR MODELER can also be found there.
Visit the StruMM3D Molecular Models and Templates page to look for new structures/templates that can be valuable for building more complex molecular models.
The StruMM3D user's guide (also the online help file) for StruMM3D is updated quite frequently and is available for download. Current users of the StruMM3D MOLECULAR MODELER should put a copy of this file into their \STR3DI directories.
The recent versions of StruMM3D have been configured to enable users of molecular databases to easily get data about their molecular models into these databases. Remember that Windows 95/98/ME/NT/XP/7/8.8.1/10 compatible database manager, DBOX32, is available, with several exciting features. The DBOX database manager is designed to enable users to create molecular databases that can contain text and interface seamlessly with StruMM3D to conduct molecular modeling. This simplifies the storage and retrieval of information about your molecular models and x-ray data, without having to tussle with paper-based files containing long lists of cryptic file names.
EXORGA's ACADEMIC PARTNERSHIP PROGRAM
Are you planning to introduce your undergraduates to molecular modeling, or already have an existing molecular modeling course/workshop in your chemistry program?
Exorga wants to get StruMM3D into every undergraduate molecular modeling course and has initiated an Academic Partnership Program that is designed to provide schools and colleges with unlimited technical support and outstanding customer service.
We'll schedule site visits by our professionals to instruct you and your students in the use of our programs. We'll show you how to get involved in exciting and intellectually stimulating molecular modeling exercises, cheaply.
E-mail us, including your name, affiliation, and a brief description of your (proposed) program, and we'll provide you with all of the information you'll need to access this valuable new service.
UPGRADE CHAT
Every now and then we rigorously test the QVBMM molecular mechanics force field. It isn't that we have found anything "wrong" with the force field, but, being perfectionists, we can't help wondering if we can make it a bit better. No major re-parameterization has ever been found to be necessary, and the changes that have been implemented are quite small, but they do result in better simulations of complex biomolecules such as DNA/RNA oligomers, sugars, peptides, etc..
Molecular modeling programs are memory hogs! They use large arrays and really grab hold of the computer’s resources. StruMM3D automatically helps you to husband your computer's precious memory resources using its Automatic Memory Management feature that enables StruMM3D to use the minimum memory required during its operations. This feature greatly facilitates multi-tasking when using StruMM3D in Windows 95/98/NT/XP/Vista/7 systems.
StruMM3D TEMPLATES
As we stated above, we do have many template structures that you can use to build even more complex molecular models in StruMM3D. We will share these data with you, if you ask, and so we have stocked a web page with the files of some of these StruMM3D template structures in order for you to test the efficacy of this offer. If you need a template, and did not find it with those you downloaded, then just drop us a line and we'll place the data file in the StruMM3D TEMPLATE STRUCTURES page.
TIPS FOR USING THE StruMM3D MOLECULAR MODELER
Our customer Service has initiated a page devoted to addressing, in a reviewable form, some of the questions they are most frequently asked. We prefer to regard this page, not as a FAQ, but as a compilation of StruMM3D Tips and Shortcuts that will ease and speed your developing acquaintanceship with our molecular modelers.
REMEMBER
Follow the Exorga's DownLoadables hyperlink to get information about obtaining the StruMM3D molecular modelers. There, you can download appropriate order/registration forms so that you can become a fully registered user of StruMM3D and get the latest versions at low cost, in addition to getting all of the additional software, manuals, and customer support.
EXORGA INC. - Consultants in ALL areas of organic chemistry and Custom chemistry software producers!
MSDOS ® © and WINDOWS ® © are programs and registered trademarks of The MicroSoft Corporation ®.