A company of consultants in chemistry and chemical engineering, actively involved in computational, experimental, and environmental organic chemistry.
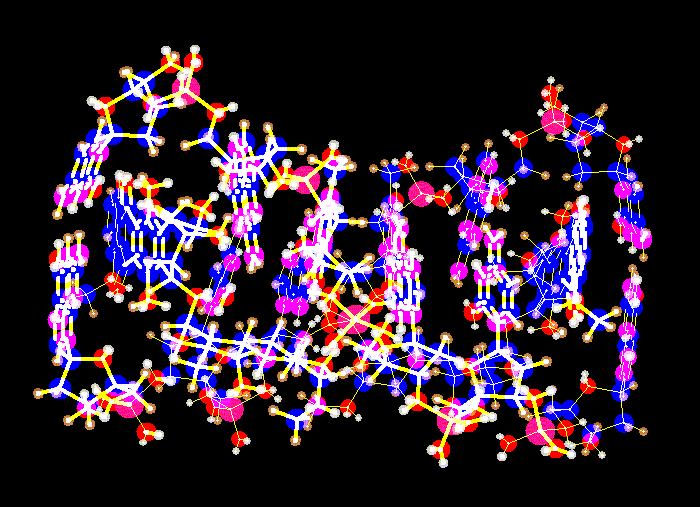
An anthraquinone intercalated into a DNA double helix, from the x-ray coordinate data.
Find out about StruMM3D's molecular models, how you can generate them, and how you can use them in your daily tasks, presentations, reports and publications.
THE STR3DI MOLECULAR MODELERS
StruMM3D generates the best molecular simulations of single molecules and docked molecular clusters, of any molecular mechanics program currently available.
The advanced structure analytical algorithms of StruMM3D are tried and tested, and shown to be vastly superior to others, in detecting subtle stereo-electronic effects embedded in diffraction generated coordinate data, like aromaticity and anomeric effects.
If you are examining diffraction (x-ray, etc) coordinate data for accuracy and reliability, then only StruMM3D’s proven and superior structure analytical algorithms will ensure you of accurate analysis.
·The QVBMM force field, the engine of StruMM3D, has been optimized for performance. The program is now compiled to produce the fastest and leanest code.
·Thereis an extensive library of template structures in thedirectory “Templates”, in \Str3Di. Whenever you wish to load a template structure, that “Template” directory is at the top of your search list. Remember that there are many structure files saved in \Str3Di\Data, and the other sub-directories, and all of these can be used as templates. Even structures from x-ray crystallography, that are stored in \Str3Di\X-ray, can be used as templates.
·One of the most powerful, but much overlooked routines for molecule construction, by modifying template structures, CONSTRUCTION - CHANGE ATOM TYPE, has been tweaked and made even better. You can change hydrogens, or any atom, to any other atom, with full retention of stereochemistry with a few click of the mouse.
·A routine for facilitating the linking of template structures into bigger, more complex, structures has been added - CONSTRUCTION - LINK TEMPLATE STRUCTURES. The routine for linking any combination of molecules onscreen into one logical structure has been appropriately renamed to - CONSTRUCTION - MAKE ONE LOGICAL STRUCTURE.
·A new utility, Str3Dinv.Exe, now prompts StruMM3D to automatically perform a complete inventory of all SXS and .XXS files in the \Str3Di directory, and to use the data to update the MOLECULE.LOG database file. If you have several thousand structure files, the way we do, and might need to refresh functional group data in the MOLECULE.LOG, then this is VERY useful.
·An improved molecular database. StruMM3D records and monitors data (including a functional group analysis) on newly simulated molecules in it’s MOLECULE.LOG database file. Invalid/outdated entries are automatically flagged and deleted. An improved method for searching the log/database is now available.
·By now you would know about the Molecular Log that StruMM3D uses to create an inventory of the molecules accessed, and this log file can be searched from withing StruMM3D to find wjatever you need. In version 8.0.0.14 we also introduced a more "visual" way to search for target molecules. If you copy the screen/displayed molecule to the clipboard a copy of the BMP file is also automatically stored in the working directory. You can use that BMP file in your publications or documents, and you can also convert it to the smaller JPG or GIF formats using third party software.
The collection of BMP/JPG/GIF files can become a visual record of your files, via their thumbnails.
If you have kept the graphics file in the same directory as the coordinate data file, then when you right click on the graphics file and select the "Open With" option to use StruMM3D to open the file, the molecular modeling exercise will be launched. So now you have a visual way of identifying molecules and loading them into StruMM3D, as well as the older Molecule Log method.
·An extended set of structure drawing tools and options. Now the structures of new molecules can be created and modified using the recently updated CONSTRUCTION - CREATE/MODIFY a MOLECULE routine, AND the structures of existing molecules can be modified using the recently updated CONSTRUCTION - ADD ATOM/H/lp routine.
·Having only one log file to record data for a molecular simulation can lead to a jumble of data. Now there are specialized logs for recording lone pair interactions, steric and torsional interactions, conformer energy data, coordinate data, and one more for all other data. Now its easier to move some of these data into spreadsheets.
·Accommodates metal atoms chelated by organic ligands, up to a coordination number of 15. Do you know of a more highly coordinated system?
·A routine for calculating the molar enthalpies of formation of simple small molecules. This routine is still under construction, and will be improved in future versions. At present, it gives good data for molecules comprised of C, H, N and O.
·A totally reworked routine for automatically importing molecules of any size, up to 10000 atoms and lone pairs. No user input is necessary to adjust the programme’s limits, etc.
·A totally new scaling algorithm for changing molecule sizes, that enables the program to automatically display molecules of all sizes, when in “autosize” mode. An optional “autozoom” feature that lets you watch the structure size adjustments.
· The Move/Measure routine automatically shows the energy of the molecule, if it is fully valence elaborated with hydrogens and lone pairs.
·The file importer FILECONV now works totally transparently, and automatically, with StruMM3D, and it is a joy. Now you can just click on any PDB, CIF, CSD, MOL, M3D, INP, XYZ, and many other file types, to be swiftly presented with the molecular simulation. Remember that StruMM3D exports MM2, MMX and XYZ files.
·StruMM3D will insert the molecule’s functional groups into the database, molecule.log, in addition to your own (VERY BRIEF) molecular description. This evolving feature will enhance your ability to search for molecules with special features.
·Atom coordinate data is saved, once again, in single precision numbers, so making the data files even smaller.
·Crystallographers and unit cell constructors will appreciate the new features.
·And, if you like sound effects, now we’ve got some. StruMM3D plays .MP3 files. Feel free to suggest, or substitute, ones you like.
Meeting More Challenges
Most C-H, O-H and N-H bonds in x-ray crystal structures are too short! In fact, based on the covalent radii of the atoms involved, in most instances the bond lengths would correspond to bond orders of 2! This is such a widespread problem that we tweaked StruMM3D to automatically handle these errors. We also tweaked the graphics to enhance the viewing of multiple bonds in molecules, and the clipboard routine to give more options in copying images.
Then, there is the totally unfathomable reluctance of the PDB group to require that element symbols be written as required in all of the rest of chemistry (Ca, Pb, Sb, etc, while PDB files use CA, PB, SB). So PDB files, as now written, can confuse users between calcium (written CA in PDB files) and carbon-alfa (also written CA in PDB files). Talk about nightmares. We’ll just have to keep making FileConv smarter, and we have, but get with the times PDB!
More Modern
StruMM3D, “the next generation of molecular modelers”, powered by the new QVBMM molecular mechanics force field, is a high-level molecular modeling tool has re-defined the standards for molecular mechanics programs. StruMM3Dwill enable you to tackle theoretical problems in organic chemistry that are outside of the reach of all other molecular mechanics based force fields. You'll also be able to explore some problems that cannot be fully resolved by the currently available molecular orbital-based methods.
For example, acetals show unusual conformational preferences that have puzzled theoreticians for many years, and this phenomenon has been called the Anomeric Effect. In 1998, following a study of saccharides and other simple acetals, using StruMM3D and data from the Cambridge Crystallographic Database, Vernon. G. S. Box published (Heterocycles) a landmark paper in which the roles of C-H hydrogen bonding, and lone pair interactions, were definitively used to rationalize the Anomeric Effect, and the Reverse Anomeric Effect. At that time, MO calculations were unable to simulate these stereo-electronic effects. It was only in 2007 that MO studies by O. Takahashi, et al., were able to corroborate the roles of C-H hydrogen bonding in the Anomeric Effect (Carbohydrate Research).
More Versatile
No substructure templates used in energy minimization
Have you ever wondered how the MM2, MM3, MM4, MMX programs manage to perform such fast structure energy minimizations? Well, these programmes use a massive set of “functional group templates” to squish the molecular model into, rather than to truly search for a minimum energy structure. So MM3 and MM4 have templates for 46 kinds of oxygens, 21 kinds of nitrogen, 16 kinds of carbon, 9 kinds of sulfur, etc. You might as well use an elaborate set of mechanical models that has 46 types oxygen atoms, 16 types of carbons, etc, and you would get the same data as that from MM3 and MM4, with less hassle.
StruMM3D has one set of parameters that are widely used for each hybridization state, for each of the common atoms, just the way nature does it. There are only two situations in which StruMM3D uses special parameters, and these are for cyclopropane/cyclobutane carbons and for thiophenes sulfurs. StruMM3D executes a thorough search for minimum energy structures, no fiddles or artificial templates are used, and the results are the most reliable. This fundamentally different type of search for structure energy minima takes a bit longer than using templates, but is much more congruent with true scientific principles, and much more reliable.
Incidentally, MM4 was released in 2003 and started to try to use polarized bonds, the way StruMM3D did seventeen * 17 * years before!
Versatile cluster creation and handling
We all know about the great importance of molecular associations and clusters. Whether we are considering simple dimers, trimers, or other oligomers, or the more complex cases of molecular clusters like solvated species. The potential energies of molecules can vary dramatically if they are associated with other atoms of molecules, as opposed to if they are isolated. The same applies to the energies of the conformations of molecules. So, it is obvious that occasionally you will wish to study molecular complexes.
StruMM3D can handle molecular complexes. Most of the other molecular mechanics programs, MM2, MM3, MM4, MMX, PCModel, and others, simply cannot minimize the structures of molecular clusters. The algorithms that these programs use to generate rapid, and sometimes erroneous, minimizations would not allow them to handle molecular clusters properly.
Easy “trapping” of high energy conformers
Have you ever wished to stop the structure minimization process at some point that is not a minimum, in order to examine the molecule’s parameters? StruMM3D allows you to do that. You can’t do this if you are using other molecular modeling programs, especially if you don’t want to place artificial geometrical constraints on your molecule.
Easy importation of data in several data file formats
The utility program FILECONV was specifically developed to convert molecular modeling file formats into those native to StruMM3D and can be executed from within StruMM3D. FILECONV can also be used as a stand-alone program. Thus, StruMM3D, in conjunction with FILECONV, will read files written in the following formats and for the following programs - Alchemy, Cambridge Crystallographic Data Centre (CIF, CMF and CSD), M3D, MDL, MM2, MM3, INP, MMX, MOL, MOL2, MOPAC, PCModel, PDB, Schakal, XYZ as well as its native files (MXS, SXS, XCC and XXS). FILECONV, can be used as a stand-alone program, and is also automatically activated from within StruMM3D.
We have seen the true range of human folly in the way people put together structure data files. Even with strong standards for files like CIF, or PDB, the guys sometimes even omit the name of the molecule under study. These errors can stymie FILECONV, and if they do, then you might need to go familiarize yourself with the proper file format, adjust the one you wish to work with, and then reuse FILECONV. Sorry about that, but until we have self-critical scientists, the problem will remain.
Better Simulations and Data
We have generated, and structure-energy minimized, molecular simulations using MM2, MM3, MMX, and PCModel, and then subjected these to structure-energy minimizations using StruMM3D. In every case, StruMM3D generated better, more geometrically accurate when compared to x-ray diffraction data, minimum energy structures.
Some people seem to be bothered by the time it takes for StruMM3D to minimize the structure energy of a molecular model. You should be aware that StruMM3D will generate a structure that is within 0.2 kcals/mol of the closest energy minimum within two minimization cycles. If you want a “quick and dirty” structure, then you can stop the minimization process at any time, or you could have used one of the other molecular modeling programs. On the other hand, if you let StruMM3D alone, it will do the most thorough search of the molecule’s potential energy surface that any molecular modeling program can do, and that takes time. Further, you should be aware that when a structure energy minimization process is being launched, you do have the option of going for a quick minimization.
Better Data Analysis
Data from diffraction studies (x-ray crystallography, etc) are only as useful as the method that converts the data into realistic molecular structural information. Often diffraction derived coordinate data have errors that are overlooked by crystallographers that use inferior molecular modeling programs, because these programs cannot draw the experimenter’s attention to these errors. StruMM3D will immediately draw its user’s attention to these errors.
The other molecular modeling methods, which rely on a user generated atom connectivity file to construct the molecular model, will often display structures that have ridiculous bond lengths, as normal structures.
These errors are not apparent in the structure unless the user of these other programmes goes measuring all of the bond lengths. There are thousands of instances of coordinate data for molecules, available on the WWW, and from the scientific literature, that have these errors. So, do you know what you are really looking at? We have a large collection of coordinate data, from all kinds of sources that show molecules with some single bonds having the lengths of triple bonds.
Other molecular modeling programs don’t attract the user’s attention to this kind of data error, but StruMM3D does. StruMM3D will always show the user a displayed structure that is consistent with the implied bond data (lengths and angles), and if there are errors, then these errors will be very visible onscreen.
More Programme Updates
We are always trying to develop new algorithms that will enable StruMM3D to generate better molecular simulations. Sometime these efforts bomb, but sometimes they work! We think that it is good science to try to develop new algorithms, since they might offer subtle insights into the way things really work.
More Technical Support
Current StruMM3D users should keep abreast of new developments in the STR3DI molecular modelers by visiting the STR3DI TEMPLATES SITE, the STR3DI UPDATE NEWS SITE., and the STR3DI NEWS SITE. When you are there, remember to check out the TIPS and HINTS help page.
We have worked on eradicating installation problems by now offering a download of a zipped folder of a “newly installed” \STR3DI directory. Many people will find this easier than downloading the installation file, even though this file offers a more versatile installation method. We do apologize for any installation inconveniences you’ve had.
Windows Vista/7/8/8.1/10 users should watch out for security issues when installing and running any new program.
For more details about StruMM3D jump to StruMM3D details and StruMM3D feature list
What Do You NEED To Do?
Accept the challenge! Run molecular simulations using StruMM3D, and then using any other molecular mechanics program that you wish, and see which program produces the best simulation. StruMM3D gives you complete control of your molecular modeling experiences.
EXORGA INC. - Consultants in ALL areas of organic chemistry and Custom chemistry software producers!
MSDOS ® © and WINDOWS ® © are programs and registered trademarks of The MicroSoft Corporation ®.